research
Research vision and projects of the Computational Imaging and Discovery Lab.
Research vision — Building cryo-EM foundation models for particle detection and structural inference
Our research is driven by a central question: how can we transform cryo-EM into a predictive, data-driven science that explains cellular mechanisms and accelerates drug discovery? To answer it, we develop computational methods that combine AI, statistical modeling, and cryo-EM physics to unlock structures that remain invisible to current pipelines.
Over the past decade, innovations in direct electron detectors (DEDs) and image processing have driven exponential growth in the biomolecular structures solved by cryo-EM. In single-particle cryo-EM, purified biomolecules are dispersed onto a support grid and rapidly frozen into a thin layer of vitreous ice. The grid is then imaged in an electron microscope to record movies — micrographs. Thousands of individual particle images representing different views are picked from these noisy micrographs, aligned, and averaged to reconstruct the high-resolution 3D structure of the macromolecule. Beyond single-particle cryo-EM, thinned cellular samples can be prepared by focused ion beam (FIB) milling and imaged via cryo-electron tomography (cryo-ET) to reconstruct the 3D organization of cellular environments and study biomolecules in situ. Since the resolution revolution in 2013, cryo-EM has solved many structures that were intractable with other methods.
Yet major challenges remain. Cryo-EM still struggles with small, flexible, or heterogeneous targets, and cryo-ET datasets remain noisy, incomplete, and difficult to interpret.
Advances in microscope hardware and image processing are required to improve signal-to-noise ratio and data throughput. Our research combines numerical analysis and deep learning to push cryo-EM/ET data processing forward. Building on the high-resolution 2D template matching (2DTM) framework, we leverage the expanding repository of high-resolution structures and AlphaFold predictions as priors to detect more challenging targets in images of diverse specimens.
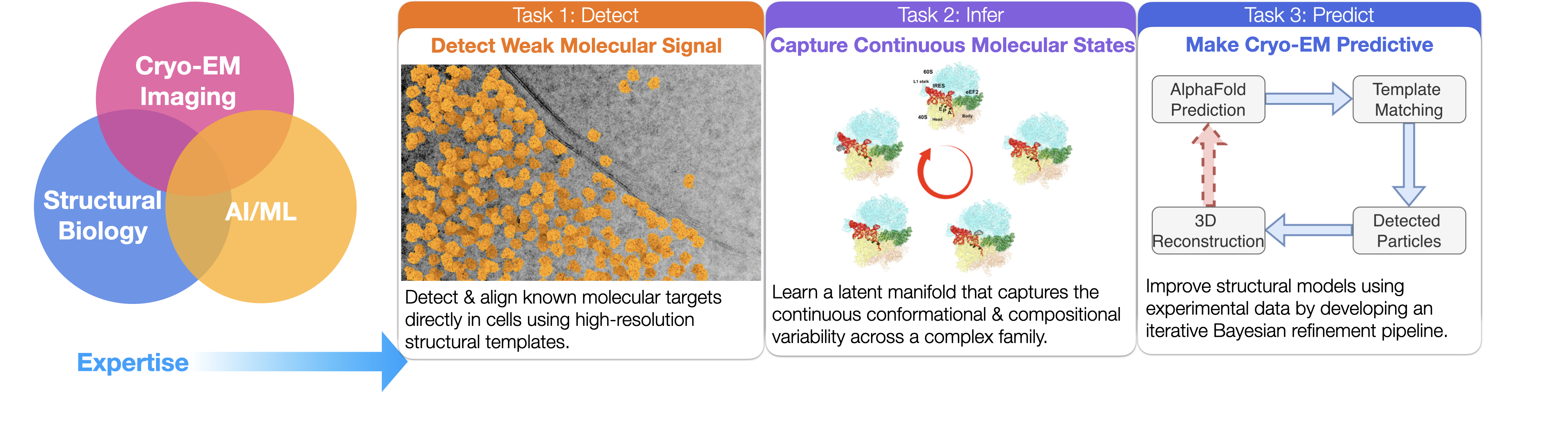
Our work lives at the interface of cryo-EM, deep learning, and structural biology. By accurately modeling the conformational states of biomolecules, we establish prior knowledge of their localization within cells — deepening our understanding of their functions and advancing structure-based drug discovery.
Current and past projects
A summary of each project below; follow the links for details and figures on the projects page.
1. DiffPose: Differentiable projection matching for fast pose inference in cryo-EM
DiffPose is a differentiable framework that uses neural networks to accelerate pose inference and generalize across diverse imaging conditions. In our experiments it achieves orders-of-magnitude faster performance than conventional grid search.
2. GisAPR: Atomic-model refinement directly against cryo-EM images
GisAPR refines atomic coordinates directly against raw cryo-EM images, bypassing the traditional 3D reconstruction step.
3. Improved cryo-EM reconstruction of sub-50 kDa complexes using 2DTM
We extend single-particle cryo-EM to the sub-50 kDa regime by leveraging 2D template matching for accurate alignment and stringent particle selection — improving visualization of ligands and binding pockets in small complexes, and laying out a roadmap to push the molecular-weight limit below 10 kDa.
4. Robust target detection with the 2DTM p-value
A new statistical metric — the 2DTM p-value — that improves detection of small or rare targets in cryo-EM, including a 193 kDa clathrin monomer, with rigorous false-positive control.
5. In situ single-particle classification
A maximum-likelihood method that probabilistically classifies cryo-EM targets detected by 2DTM with multiple templates — the first demonstration of in situ single-particle classification without 3D reconstruction. Applied to study the 60S ribosome biogenesis pathway in FIB-milled yeast cells.
6. Multiscale RNA modeling with NMR chemical shifts
PhD work developing CS-Fold, CS-BME, and CS-Annotate — computational frameworks that use NMR chemical shifts to predict RNA secondary structure, model conformational ensembles, and annotate RNA structural models.